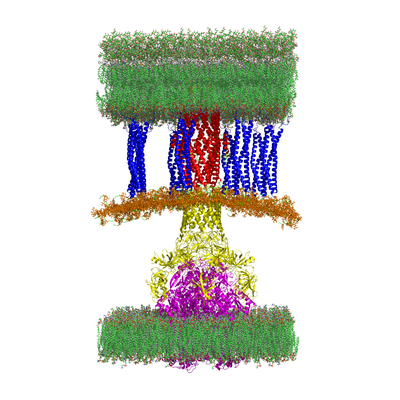
The problem of antibiotic resistance is an emerging public health threat. Many bacteria are resistant to not just one antibiotic, but many different antibiotics. In fact, some bacteria are resistant to all currently available antibiotics. One of the primary causes of resistance in bacteria are multiprotein complexes known as efflux pumps. When an antibiotic enters a bacterial cell, the efflux pump kicks it back out preventing the antibiotic from fulfilling its function of killing bacteria and healing the infection. At CMB in collaboration with JC Gumbart at Georgia Tech, Helen Zgurskaya at Oklahoma University, and John Walker at St. Louis University School of Medicine, we are using three-dimensional models of each component and the full complex of the efflux pump in Escherichia coli to discover new small molecules that can inhibit the pump, design new molecules from discovered scaffolds, and gain an atomistic understanding of the assembly and transport mechanism.
Publications:
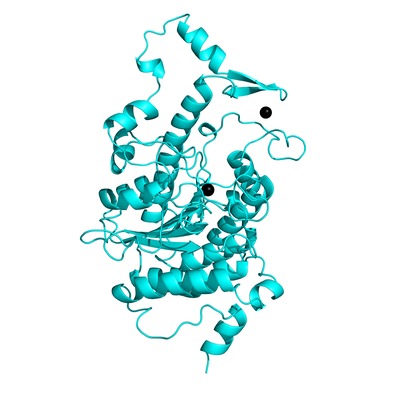
Histone deacetylase 4 (HDAC4) is a member of the histone deacetylase family of enzymes and is believed to function as a scaffold for the recruitment of other multiprotein complexes like Nuclear receptor co-repressor (NCoR). However, it has been reported that HDAC4 is overexpressed in esophageal squamous cell carcinoma (ESCC) and prostate cancer cells. All the known HDAC4 inhibitors till date are non-selective binders and cause toxicity. Hence to effectively prevent HDAC4 activity in cancer growth, there is a need to identify selective inhibitors. In our study, we have used a novel strategy to inhibit HDAC4 by targeting the interface of HDAC4-NCoR instead of the catalytic pocket. Targeting a protein-protein interface (PPI) has been challenging because in most cases, they lack cavities for small molecules to bind or are intrinsically disordered. However, using molecular dynamics (MD) simulations, We have identified novel cavities at the PPI which were not evident in the crystal structures. To find potential inhibitors, we have performed a high-throughput virtual screening of small molecules to target these new pockets.

Infective Endocarditis (IE). IE is a life-threatening cardiovascular infection associated with significant morbidity and mortality. We are working on adhesin proteins that mediate microbe attachment to human platelets via sialoglycans thus enabling pathogenesis. IE kills thousands of people in the United States alone but there are no known anti-adhesion molecules to target the disease early. Our major goal is thus to find small molecules which inhibit by binding at the sialoglycan binding pocket (competitive inhibitors) or bind to an allosteric pocket (allosteric inhibitors). We have performed first-round of virtual screening to identify novel competitive inhibitors which will be tested by our collaborators at Vanderbilt University.
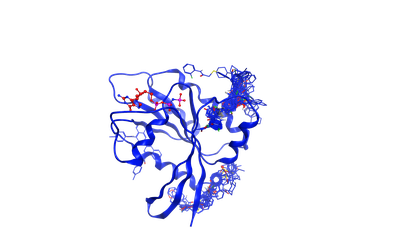
Normal KRAS proteins have a vital role in cell division, differentiation and in tissue signaling. However, dysfunctional KRAS proteins can lead to the development of malignant tumors like colorectal and lung cancers. Despite its role in cancer development, there is no effective drug against KRAS at present. Hence, there is a need to perform a high-throughput study to identify novel KRAS inhibitors. On this project, The work is done by Rupesh Agarwal, Wade Seifert and Dr. Micholas D. Smith. We want to identify novel scaffolds of small molecules which bind to allosteric sites of KRAS rather than at native substrate binding site. Our plan is to perform a high-thoughput virtual screening of 16 million small molecules present in the ZINC database on an ensemble of KRAS structures obtained from MD simulations and use this to identify a common pharmacophore or scaffold between the hits.
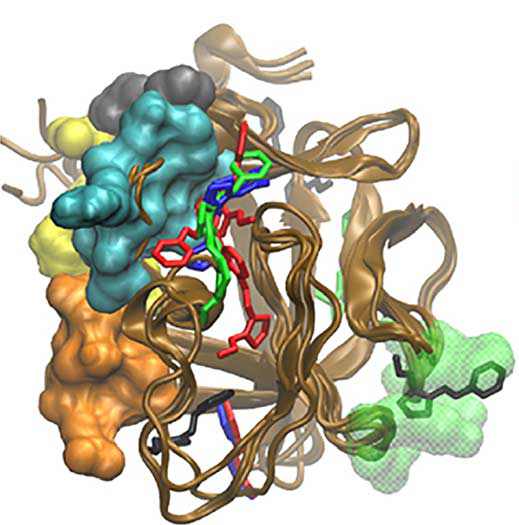
Patients with excess fibroblast growth factor-23 (FGF-23) in their circulation have metabolic deficiencies that contribute to chronic kidney disease and the bone-softening disorder rickets. We used computationally based screening (ensemble docking and virtual high-throughput screening) to identify a compound predicted to bind the FGF-23. When tested experimentally, the compound was selective for FGF-23 compared to other FGF family ligands and prevented FGF signaling in cultured kidney cells.
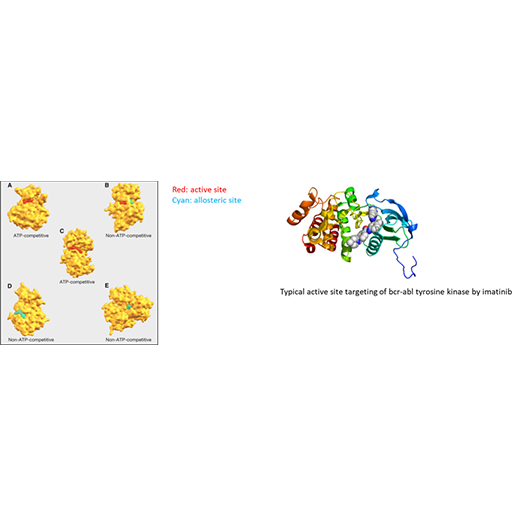
Developing a quantitative understanding of the structure, function, and dynamics of protein-protein, protein-ligand, and protein-nucleic acid interactions, and how such interactions modulate protein function, is at the heart of a molecular-level understanding of disease and how to target mutant or otherwise pathogenic proteins with drugs that have a low side-effect profile. Towards this end, computational strategies to predict high-specificity binding of drug ligands to proteins and strategies to simulate how the binding of such ligands modulates protein function are being developed, with a specific focus on targeted cancer therapy as a replacement for chemotherapy. Specifically, machine learning methods using a range of physicochemical features will be applied to the development of highly accurate scoring functions for prediction of drug ligand binding sites. After this stage, advanced sampling methods such as the Markov state model and features calculated therefrom, such as mutual information among protein residues, or direct observation of a modulatory effect, will be used to assess allostery, that is, the ability of a binding event at one site to modulate the active site function. Targeting allosteric sites holds the promise of greater specificity and a lower side effect profile compared with the standard approach of targeted cancer therapy involving inhibition of the active site.