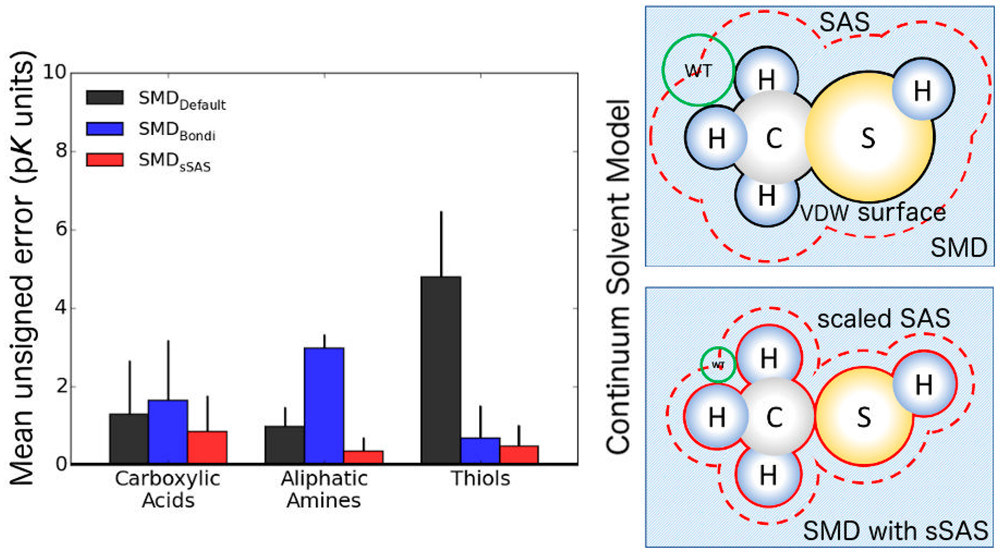
Developing accurate quantum chemical approaches for calculating pKas is of broad interest. Useful accuracy can be obtained by using density functional theory (DFT) in combination with a polarizable continuum solvent model. However, some classes of molecules present problems for this approach, yielding errors greater than 5 pK units. Various methods have been developed to improve the accuracy of the combined strategy. These methods perform well but either do not generalize or introduce additional degrees of freedom, increasing the computational cost. The Solvation Model based on Density (SMD) has emerged as one of the most commonly used continuum solvent models. Nevertheless, for some classes of organic compounds, e.g., thiols, the pKas calculated with the original SMD model show errors of 6–10 pK units, and we traced these errors to inaccuracies in the solvation free energies of the anions. To improve the accuracy of pKas calculated with DFT and the SMD model, we developed a scaled solvent-accessible surface approach for constructing the solute–solvent boundary. By using a “direct” approach, in which all quantities are computed in the presence of the continuum solvent, the use of thermodynamic cycles is avoided. Furthermore, no explicit water molecules are required. Three benchmark data sets of experimentally measured pKa values, including 28 carboxylic acids, 10 aliphatic amines, and 45 thiols, were used to assess the optimized SMD model, which we call SMD with a scaled solvent-accessible surface (SMDsSAS). Of the methods tested, the M06-2X density functional approximation, 6-31+G(d,p) basis set, and SMDsSAS solvent model provided the most accurate pKas for each set, yielding mean unsigned errors of 0.9, 0.4, and 0.5 pK units, respectively, for carboxylic acids, aliphatic amines, and thiols. This approach is therefore useful for efficiently calculating the pKas of environmentally relevant functional groups.
Peng Lian, Ryne C. Johnston, Jerry M. Parks, and Jeremy C. Smith. J. Phys. Chem A, 2018, 122(17), pp 4366-4374
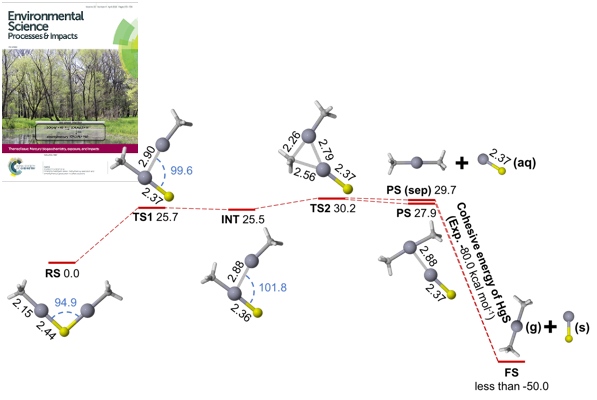
Decades of research have been devoted to understanding methylmercury formation and degradation, but little is known about the formation of dimethylmercury in aquatic systems. In collaboration with Andrew Graham at Grinnell College and Niri Govind at the Environmental Molecular Sciences Laboratory (EMSL) at Pacific Northwest National Laboratory (PNNL), we combined complementary experimental and computational approaches to examine methylmercury speciation and dimethylmercury formation in sulfidic solutions. We found that bis(methylmercuric) sulfide, (CH3Hg)2S, decomposes slowly to form dimethylmercury and mercury sulfide (HgS). Using relativistic quantum chemical calculations, we proposed a mechanism for the reaction that is in excellent agreement with the experimental kinetics measurements. In natural systems with relatively high methylmercury/sulfide ratios (e.g., at the oxic/anoxic interface), highly toxic dimethylmercury may be produced.
Kanzler, C.R., Lian, P., Trainer, E.L., Yang, X., Govind, N., Parks, J.M. and Graham, A.M. Environ. Sci.: Processes Impacts 2018, 20, 584-594.
Peng Lian, Ryne C. Johnston, Jerry M. Parks, and Jeremy C. Smith. J. Phys. Chem A, 2018, 122(17), pp 4366-4374
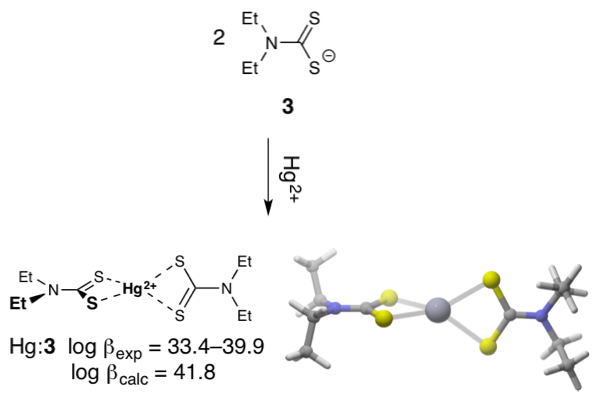
The chemical speciation and bioavailability of mercury (Hg) is markedly influenced by its complexation with natural dissolved organic matter (DOM) in aquatic environments. To date, however, analytical methodologies capable of identifying specific Hg-DOM complexes are scarce. In collaboration with Baohua Gu, who led the work, we identified candidate molecules with the observed chemical formula (C8H17N2S4Hg+) and then used density functional theory (DFT) to predict their Hg-binding properties and solution-phase structures. Our prediction that the most prevalent complex is a HgII bis(N,N-diethyldithiocarbamate) complex was subsequently verified with mass spectrometry experiments.
Chen, H., Johnston, R.C., Mann, B.F., Chu, R.K., Tolic, N., Parks, J.M. and Gu, B. Environ. Sci. Technol. Lett. 2017, 4, 59–65.
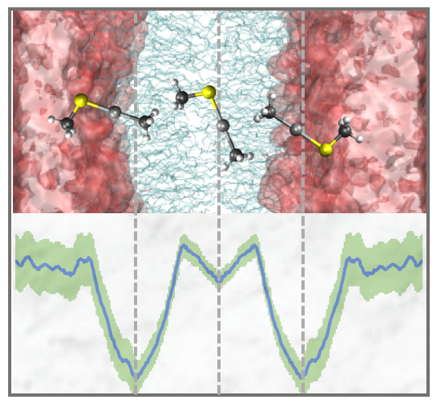
Cellular uptake and export are important steps in the biotransformation of mercury by microorganisms. However, the mechanisms of transport across biological membranes remain unclear. To identify biophysical features that govern Hg uptake, we performed extensive molecular dynamics simulations of the passive permeation of HgII and methylmercury complexes with thiolate ligands through a model bacterial cytoplasmic membrane. We found that favorable interactions with carbonyl and tail groups of phospholipids stabilize Hg-containing solutes in the tail–head interface region of the membrane.
Zhou, J., Smith, M.D., Cooper, S.J., Cheng, X., Smith, J.C. and Parks, J.M., Environ. Sci. Technol., 2017, 51, 10595–10604.
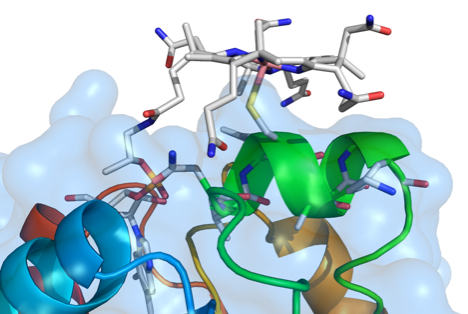
We collaborated with Judy Wall’s group at the University of Missouri, who performed site-directed mutagenesis and protein sequence truncation, to determine amino acid residues in HgcA and HgcB that are important for mercury methylation. Mutations of conserved amino acids near the strictly conserved Cys residue, Cys93, in HgcA had varying impacts on mercury methylation capacity but showed that the structure of the putative ‘cap helix’ region harboring Cys93 is crucial for methylation. In the ferredoxin HgcB, only one of two conserved cysteines at the C-terminus is necessary for methylation. An additional, strictly conserved Cys in HgcB was found to be essential for methylation. This study supports the previously predicted importance of Cys93 in HgcA for methylation of mercury and reveals additional residues in HgcA and HgcB that facilitate the production of this neurotoxin.
Smith, S.D., Bridou, R., Johs, A., Parks, J.M., Elias, D.A., Hurt Jr., R.A., Brown, S.D., Podar, M. and Wall, J.D. Appl. Environ. Microbiol, 2015, 81, 3205–3217.
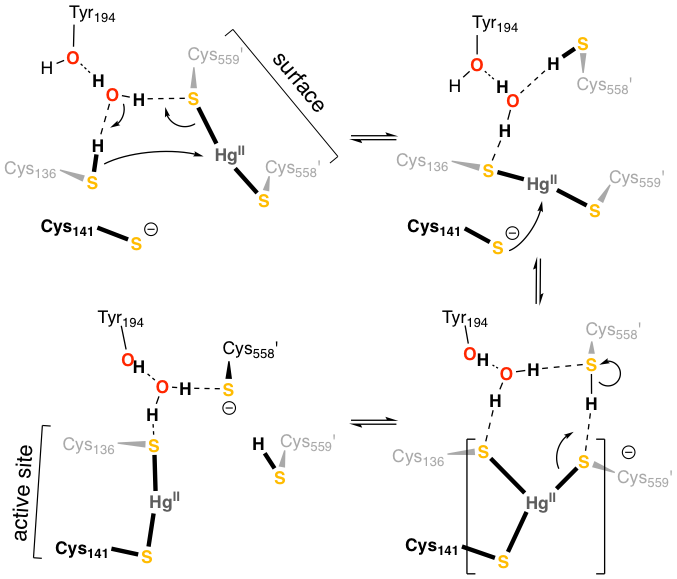
In collaboration with Sue Miller at UCSF, we combined X-ray crystallography and QM/MM simulation to understand how the bacterial enzyme mercuric ion reductase, MerA, shuttles Hg2+ ions into its active site, where it is reduced to less toxic Hg0. We found that Hg2+ transfer in MerA involves a balanced competition between Hg2+ and H+ for pairs of cysteine thiolates. Our simulations revealed at atomic detail how MerA, a key component of bacterial mercury resistance, orchestrates the handling of toxic Hg2+ through successive binding by and transfer among pairs of cysteines.
Lian, P., Guo, H.-B., Riccardi, D., Dong, A., Parks, J.M., Xu, Q., Pai, E.F., Miller, S.M., Wei, D.-Q., Smith, J.C. and Guo, H. Biochemistry, 2014, 53, 7211–7222.
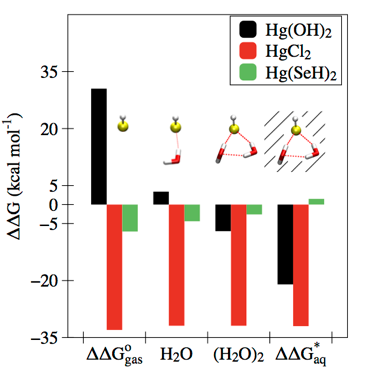
Defining the factors that determine the relative affinities of different ligands for Hg2+ is critical to understanding its speciation, transformation, and bioaccumulation in the environment. We used quantum chemistry to dissect the relative binding free energies for a series of inorganic anion complexes of Hg2+. By comparing Hg2+−ligand interactions in the gaseous and aqueous phases, we showed that differences in interactions with a few, local water molecules leads to a clear periodic trend within the chalcogenide and halide groups and recovers the well-known experimentally observed preference of Hg2+ for soft ligands such as thiols. This approach established a strong basis for understanding Hg speciation in the biosphere with quantum chemical calculations.
Riccardi, D., Guo, H.-B., Parks, J.M., Gu, B., Summers, A.O., Miller, S.M., Liang, L. and Smith, J.C. J. Phys. Chem. Lett., 2013, 4, 2317−2322.
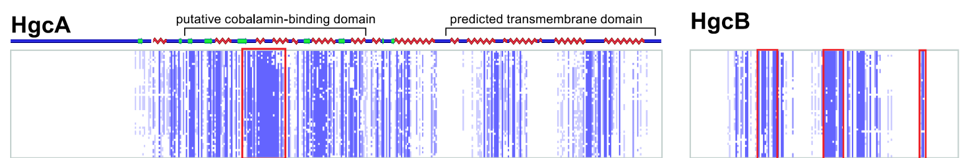
Methylmercury is a potent neurotoxin produced in natural environments from inorganic mercury by anaerobic bacteria. However, the genes and proteins responsible remained elusive for decades. We discovered a two-gene cluster, hgcAB, required for mercury methylation by Desulfovibrio desulfuricans ND132 and Geobacter sulfurreducens PCA. In either bacterium, deletion of hgcA, hgcB, or both genes abolishes mercury methylation. These genes encode a corrinoid protein, HgcA, and a ferredoxin, HgcB, consistent with roles as a methyl carrier and an electron donor required for corrinoid cofactor reduction, respectively. Among bacteria and archaea with sequenced genomes, gene orthologs are present in confirmed methylators but absent in nonmethylators, suggesting a common mercury methylation pathway in all methylating bacteria and archaea sequenced to date.
Parks, J.M., Johs, A., Podar, M., Bridou, R., Hurt Jr., R.A., Smith, S.D., Tomanicek, S.J., Qian, Y., Brown, S.D., Brandt, C.C., Palumbo, A.V., Smith, J.C., Wall, J.D., Elias, D.A. and Liang, L. Science, 2013, 339, 1332–1335.
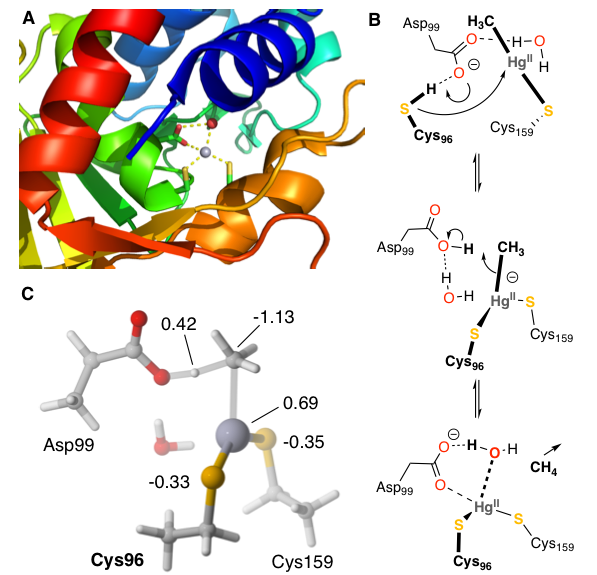
The bacterial organomercurial lyase, MerB, catalyzes the demethylation of methylmercury. Using a DFT-based quantum chemical cluster approach, we investigated two proposed reaction mechanisms of MerB. The calculations ruled out a direct protonation mechanism in which a cysteine residue delivers a catalytic proton to the methyl leaving group. Instead, the calculations support a two-step mechanism in which two Cys thiolates coordinate methylmercury and an Asp protonates the methyl leaving group to cleave the Hg-C bond and release CH4. Natural population analysis revealed that MerB lowers the activation free energy by redistributing electron density into the leaving group and away from the catalytic proton.
Parks, J.M., Guo, H., Momany, C., Liang, L., Miller, S.M., Summers, A.O. and Smith, J.C., J. Am. Chem. Soc., 2009, 131, 13278–13285.